|
Data Integration for Systems
Biology using the ONDEX system Leucorea, |
ONDEX Visualization ToolKit (OVTK) User Tutorial

Tutor:
Catherine Canevet
Rothamsted Research, Biomathematics and Bioinformatics
TABLE OF CONTENTS
1.1 OVTK
Layout and User Interface
2 Loading
Biological Networks into OVTK
2.1 Loading
Existing Biological Networks into OVTK..
2.2 Creating
an empty network and manually adding concepts and relations
4 Constructing
Biological Networks in Ondex
4.2 Ondex
back-end Workflows in XML
5 Customising
Visualisation of Biological Networks
5.1 Application
case 1: Analysing Experimental Data in the context of Integrated Biological
Networks
5.2 Application
case 2: Functional GO Annotation
5.3 Application case 3: Cooking with Protein-protein
interaction networks
5.3.3 Data sources and processing workflow
1
Introduction
to OVTK
This section will introduce
the Ondex Visulalization ToolKit user interface. A network consists of
genes/proteins/metabolites as concepts and interactions represented as links
i.e. relations between concepts.
First of all, we will look
at the basic user interface of OVTK. Then we will load up a network to show all
menu features of OVTK and some of the core functionality such as layout
algorithms, annotators and filters.
1.1
OVTK
Layout and User Interface
Launch OVTK[1]
- You should see a window that looks
like this:

- At the top of the OVTK window there is a
toolbar which contains the menus, icons and search bar. The name of each
icon is shown when the mouse pointer hovers over it.
- The rest of the window is used to show
visualization windows and pop-up interactive boxes to set up filters,
annotators, etc.
- When the OVTK is started up, a wizard pops
up in the middle of the main window. This wizard window gives a small
description of OVTK, links to two webpages (the left-hand side one is a
link for users, the right-hand side one for developers), as well as three
buttons (that are shortcuts to opening an empty network, opening an
existing network and starting the tutorial). Finally, at the bottom of the
window, users are given the option of unticking a box if they do not wish
for this wizard window to pop up automatically next time the tool is launched.
- At the bottom of the OVTK window, there is
a command line interface. Typing “help;” provides users with a manual for
it.
It is possible to undock
the top toolbar of the OVTK by a simple “drag and drop”. When this toolbar is
undocked, it is possible to re-dock it by clicking on its far left side and
again dragging and dropping to its original position.
1.2
Loading
Networks
We will now load a network
to show all features of OVTK user interface.
- Go to File->
Open from file
- You should see an Open File Dialog
- Open the tutorial/main_part folder, select aracyc.xml.gz and click on open
You should see the
following:
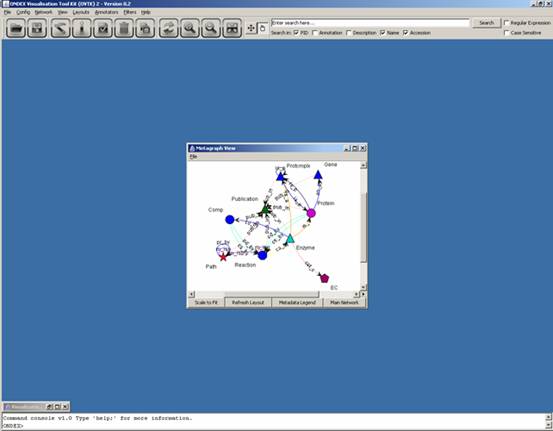
The first window that opens
is the metagraph view. Note that the main network is minimized in the bottom
left-hand side corner. The metagraph gives an overview of all the different
types of concepts present on the main network, as well as all the different
types of relations which link them.
The metagraph view has four
buttons at the bottom of its window:
- Scale to Fit: after resizing the window,
will scale the metagraph to fit it
- Refresh Layout: will draw the metagraph
again should users have moved anything and will end with a “Scale to Fit”
- Metadata Legend: will pop up a window which
displays a modifiable colour/shape legend as well as the number of
concepts and relations in the network for concept classes, controlled
vocabularies and relation types
- Main Network: will open the main network
visualization window which was minimized until then
Exercise
Trying to understand the
metagraph before opening the main network usually helps. The main network is
opened with the “Circular” layout which is a circular arrangements of all the
concept classes in the network.
Moving concepts around in
the metagraph should help you to make sense of it. This is an example of how
the metagraph can be laid out:

A protein is encoded by a
gene (en_by).
A protein is part of a
protein complex (is_p).
A protein is consumed by or
produced by a reaction (cs_by, pd_by).
An enzyme is a protein or a
protein complex (is_a).
An enzyme has a catalysing
class EC (cat_c).
An enzyme is co-factored
(or activated) by a compound (co_by).
A compound is consumed by
or produced by a reaction (cs_by, pd_by).
A reaction is a member, is
part of a pathway (m_isp).
A reaction is catalysed by
an enzyme (ca_by).
Finally, most concepts can
be mentioned in publications or publicated in (pub_in).
Right-clicking on
concepts/relations on the metagraph shows the number of concepts/relations of
that type in the network. It also gives users the opportunity to untick
“Visible”. This will make concepts/relations of that type invisible on the main
network. The concept/relation in the metagraph will appear in a paler colour.
Note: Making concepts
invisible in the network will not delete them from the network altogether. If
you wish to do so, use Network ->
Synchronise Network. A window will pop up to ask you for confirmation.
Exercise
In the metadata legend,
clicking on colours/shapes will allow you to pick different colours/shapes for
concept classes, controlled vocabularies and relation types (see three tabs).
In order to see it take effect, refresh the metadata (bottom button in the
metadata legend) and click on use View
-> Update Display.
For example, a reaction is
of the same colour as three other concept classes (gene, protein complex and
compound). In order to make it more distinct, we are going to change its colour
by clicking on the coloured rectangle (first column). We get the following
window popping up:
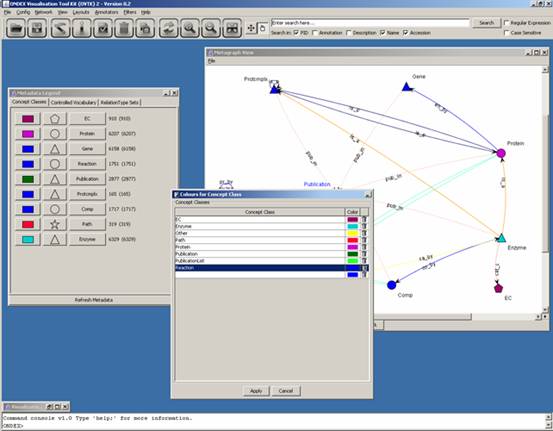
Clicking on the coloured
rectangle in the new window will pop open a “Pick a colour” window:

Select a colour, click on
OK, Apply, Refresh Metadata and View
-> Update Display to finally get the following:

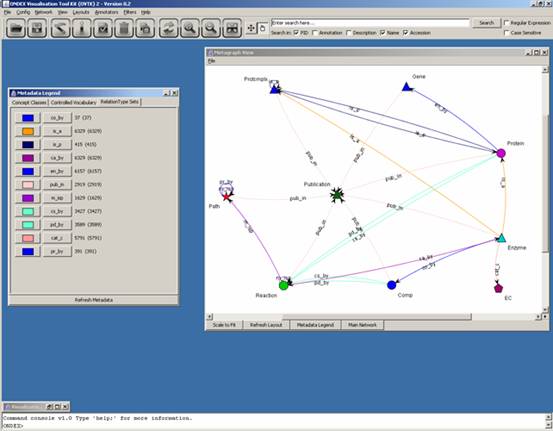
The metadata legend has
three tabs: Concept Classes, Controlled Vocabulary and RelationType Sets.
“ca_by” was originally in yellow and therefore not very visible. In the
following screenshot its colour was changed to purple:
To move on to the main
network, you can click on “Main Network” in the “Metagraph View” window or
maximize the visualization window that has been sitting in the bottom left-hand
side corner all along.
Opening the visualization
window can take a while when there are a lot of concepts and relations to draw.
Nevertheless, you will obtain something like this:
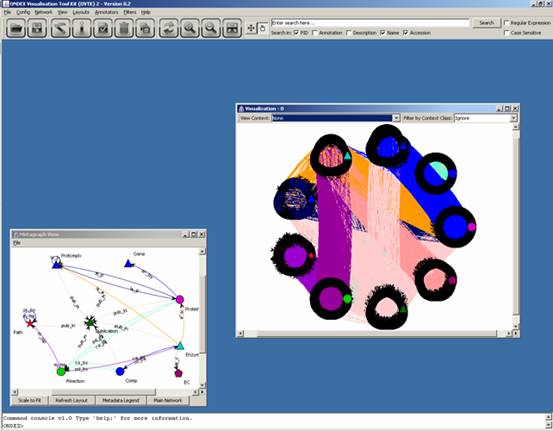
Rather than looking at this
overwhelming graph, let us select a single pathway to analyse: GDP-L-fucose biosynthesis
II
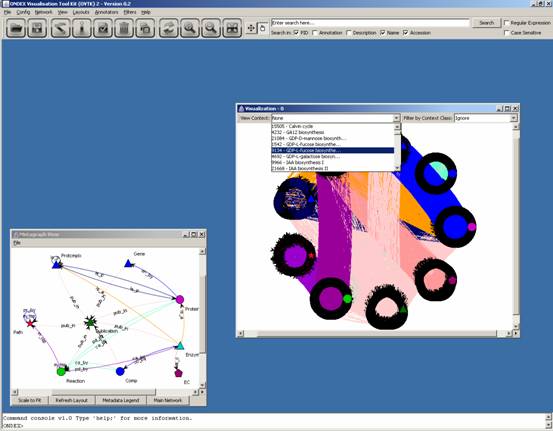
This leaves us with the GDP-L-fucose biosynthesis
II (from L-fucose) pathway only:
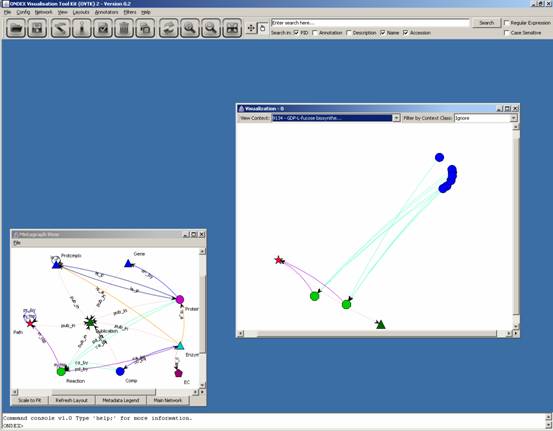
You can use Layouts → Hierarchical for OVTK to organise the concepts hierarchically:

You can then select a
concept (for example the pathway itself) and click on the “i” icon (a tooltip
shows when you mouse over the icon) to see information about it:
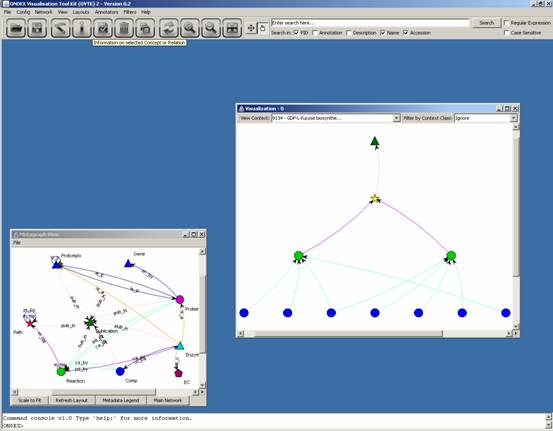
The information gives a
link to a website:
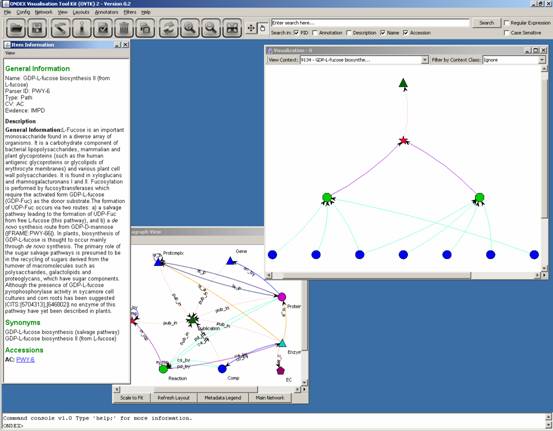
Clicking on this link opens
a web browser at the following page:
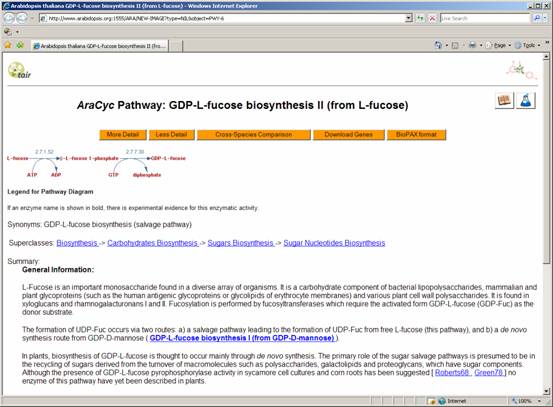
Exercise
Study the reaction shown in
the website and try to understand how it is represented in OVTK.
Help: The first thing you
might want to do is use the View menu to add concept and relation labels on the
network so you know what is what without needing the “Item Information” window.
Use
- View → Show Concept Labels
- View → Show Relation Labels
Help: To move concepts around,
- you can either pick them (in picking mode –
last icon before search bar in the toolbar) and move them one by one
- you can press shift to select a few nodes
with the left mouse button and using the left mouse button again you can
drag and drop these group of nodes
Following this process,
you obtain:

Note: the “Metagraph View”
window has been minimized.
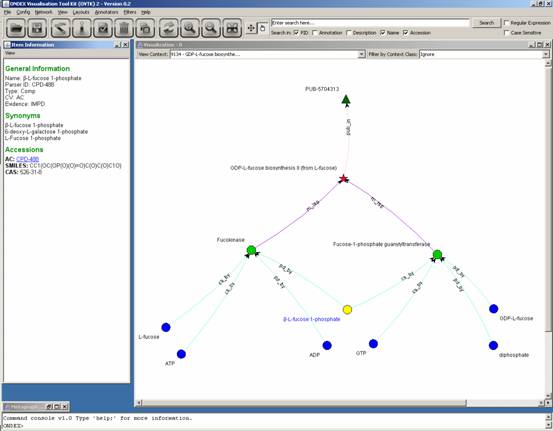
In the above screenshot, the
reactions previously shown on the webpage have been reconstituted. Here, the compound
that is a product of the first reaction and a substrate of the second has been
selected and is automatically shown in the “Item Information” window. Note:
SMILES stand for “simplified molecular input line entry specification”.
Note: If you have closed
the Metadata Legend (previously
launched from the metagraph view window), you may launch it again using Config -> Metadata Appearance.
1.3
Manipulating
Networks
Zooming
OVTK provides two
mechanisms for zooming:
i)
Using
the mouse –move the mouse scroll forward
and backwards
ii)
Using
the two zoom buttons on the toolbar
to zoom in and out of the network
Selecting an area to look at from the overview
When studying large graphs,
using the Network -> Satellite View
to move and navigate across the network can be very useful.
Manually rearrange a network
Select the concepts of
interest and move them around. See exercise in Section 1.2.
Automatically rearrange a network
There are a variety of
layout algorithms which can be found in the Layouts menu. Select Layout -> OVTK Layouts (the GEM Algorithm is probably the most
popular layout).
Rotating and Sheering your network
Finally you can rotate and
scale your network within OVTK. In the transformer mode (See Section 1.4 for icon), shift + mouse will rotate, control + mouse
will sheer.
1.4
OVTK
icons
The toolbar
is composed of several icons followed by a search bar which is explained in
further details in Section 3.1. The icons are as follows:
![]()
![]() Open
Open
![]() Save
Save
![]() Add a Concept or Relation
Add a Concept or Relation
![]() Information on selected Concept or Relation
Information on selected Concept or Relation
![]() Edit selected Concept or Relation
Edit selected Concept or Relation
![]() Delete selected Concept or Relation
Delete selected Concept or Relation
![]() Copy whole Network as new
Copy whole Network as new
![]() Refresh Layout
Refresh Layout
![]() Zoom In
Zoom In
![]() Zoom Out
Zoom Out
![]() Center Network
Center Network
![]() Set mouse to “transforming” mode (to move
within the network)
Set mouse to “transforming” mode (to move
within the network)
![]() Set mouse to “picking” mode (to select
concepts, drag and drop them)
Set mouse to “picking” mode (to select
concepts, drag and drop them)
1.5
OVTK
menus
We will
briefly run through all the menus available in OVTK.
File
The File menu contains
basic file functionality:
- File → New empty network
creates a new network
- File → Open from file
opens an OVTK session file
- File → Save to file
saves a session file
- File → Print
prints the visualization window
- File → Export as…
exports the visualization window
- File → Import…
imports files in Network WorkBench format
- File → Launcher
creates workflows for data integration (see Section 4.1)
- File → Exit
exits the OVTK
Config
The Config menu contains:
- Config → Settings
for login user and password
- Config → Metadata Appearance
to modify colours/shapes of conepts/relations
- Config → Change Concept Label Font
to change concept label font
- Config → Change Relation Label Font
to change relation label font
- Config → Save Appearance
to save the coordinates, colours and shapes of the
concepts/relations
Network
The Network menu contains:
- Network → Concept List
for a list of all the concepts
- Network → Relation List
for a list of all the relations
- Network → Satellite View
for a movable overview of the network
- Network → Metagraph View
Displays a metagraph containing all the types of
concepts/relations
- Network → Item Info
for a permanent window displaying information on
selected items of the network
- Network → Statistics
displays statistics about the network (see Section 5.3)
- Network → Center Network
centers the network within the visualization window
- Network → Synchronise Network
removes invisible items from the network
View
The View menu contains:
- View → Show Concept Labels
shows names of concepts on the main network
- View → Show Relation Labels
shows names of relations on the main network
- View → Colour Concepts by Controlled Vocabulary
colours concepts by controlled vocabularies
- View → Colour Concepts by Concept Class
colours concepts by type of concept
- View → Parse Concept Colour from General Data
Store
updates the network with information previously saved
about concept colour
- View → Parse Concept Shape from General Data
Store
updates the network with information previously saved
about concept shape
- View → Parse Relation Colour from General Data
Store
updates the network with information previously saved
about relation colour
- View → Update Display
to update the visualization window with recent
modifications
- View → Anti-aliased
smoothes relations in the network
Layouts
The Layouts menu offers a
collection of algorithms for visually organising the network:
- Layouts → Circular
for a circular arrangement of concept classes
- Layouts → Hierarchical
for a hierarchy of concept classes
- Layouts → Flip
for a flip around of selected concepts (mirror effect)
- Layouts → Kamada-Kawai
for the Kamada-Kawai layout algorithm[2]
- Layouts → Force Directed
for a force-directed algorithm based on attributes
values
- Layouts → GEM Algorithm
for the GEM layout algorithm[3]
- Layouts → Radial Tree
for a layout determined by the selection of a focus
concept
- Layouts → RelationType specific
for a force-directed algorithm based on relation types
- Layouts → Static
for the latest saved layout
- Layouts → Sugiyama
for the Sugiyama layout algorithm (“Visualization of structural information: Automatic
drawing of compound digraphs”, 1991, by Sugiyama, K. and Misue, K.)
- Layouts → Tree
for a directed rooted tree
- Layouts → Layout Options
for options on the parameters of some algorithms (some
algorithms do not have options, others do but are not yet supported)
- Layouts → Refresh layout on resize
tick for automatic relayouting of the network when the
visualization window’s size is modified
Annotators
The Annotators menu offers
a collection of algorithms for visually organising the network:
- Annotators → Betweenness Centrality
to resize concepts based on their centrality within
the network[4]
(see Section 3.3)
- Annotators → Colour by Value
to colour concepts based on one of their attribute’s
value (see Section 5.4)
- Annotators → Scale Concept by Value
to resize concepts based on one of their attribute’s
value (see Section 5.1)
- Annotators → Scale Relation Width by Value
to resize edges width based on one of their
attribute’s
- Annotators → Shape by Value
to shape concepts based on one of their attribute’s
value (see Section 5.4)
- Annotators → Virtual Knock-Out
to remove one concept at a time virtually and
calculating the structure changes in the network
Filters
The Filters menu offers a
collection of algorithms for visually organising the network:
- Filters → All Pairs Shortest Path
computes the shortest paths between all possible pairs
of concepts and removes all relations that
are not part of these shortest paths
(see Section 5.3 for an example)
- Filters → Concept Class
filters out some particular class of concepts
- Filters → Data Source
filters out concepts from a specified data source
- Filters → Neighbourhood
to see a particular number of neigbours only to a
particular concept (see Section 5.2 for an example)
- Filters → Relation Type Set
filters out some particular type of relations
- Filters → Shortest Path
computes the Dijkstra shortest path algorithm from a
particular concept (see Section 3.3 for an example)
- Filters → Significance
selects a threshold for one of the concepts
attributes’ value (see Section 5.2 for an example)
- Filters → Unconnected
filters out all unconnected concepts
Help
The Help menu offers a
collection of algorithms for visually organising the network:
- Help → About
a brief message about the project
- Help → Documentation
a documentation of OVTK with screenshots
- Help → Tutorial
this tutorial is available in html format through this
menu
Exercise
Filters and Annotators will
be further explored later on in this tutorial. For now, take some time to
familiarise yourself with the toolbar and menus that have been introduced in
the last two subsections. Play around with the currently opened network and
please ask any questions you may have.
2
Loading
Biological Networks into OVTK
In this section we will
show how to load in your own networks and associated data into OVTK. It is
possible to open biological networks into OVTK by:
- Importing network files.
- Creating an empty network and manually
adding concepts and relations.
2.1
Loading
Existing Biological Networks into OVTK
OVTK can read files written
in the following formats:
- oxl (Ondex XML, biological networks created
using Ondex)
- nwb (NetWork Bench)
- SBML, in development
- BioPax, in development
Exercise
For each concept/relation, right-click and view/edit properties.
Note: In the “Item
Information” window, you can only view information and cannot modify anything.
2.2
Creating
an empty network and manually adding concepts and relations
It is also possible to
create new, empty networks that concepts and relations can be manually added to. To
create an empty network, go to File → New
empty network. Click on the icon
for “Add a concept or relation”.
Exercise
If you wish to add a
concept, click on “Add a new Concept”. You will need to fill in all the fields
highlighted in orange as they are compulsory. This will require creating
Controlled Vocabularies, etc.
If you wish to add a
relation, click on “Add a new Relation”. You will need to select concepts
beforehand (shift + left mouse button to create a select area) so the drop-down
lists offer these concepts as origin/target of the relation.
2.3
Saving
Sessions
You can save your OVTK
sessions and export visualizations.
- To save a session, go to File -> Save to File
- To export images from OVTK visualization
window, go to File -> Export as…
3
Analysing
Networks
Searching and filtering networks is another powerful
feature of OVTK. In this section we will cover different ways of navigating and
analysing networks.
3.1
Search
OVTK includes a Search feature, which enables you to
quickly find concepts
and relations.
![]()
You may specify in which field(s) you wish to search:
parser identifier (PID), annotation, description, name and accession.
If you select (single click) one item in the search
results, you will need to single click on the icon “Zoom In” in order for OVTK
to zoom in on this particular concept. You can then use the mouse scroll to get
to the concept.
If you select several items (using the Control key) in
the search results, OVTK will automatically zoom in to show all those items as
close as possible.
Configuring
an OVTK Search
At the end of the toolbar, there are two options that
can be configured for each search.
- If you tick “Regular Expression”, you may enter a java regular
expression. An example would be: p\\d{1,4}. It will match a “p” followed by 1 to 4 digits. For more
information on regular expressions in java, please visit http://java.sun.com/docs/books/tutorial/essential/regex/
- If you tick “Case Sensitive”, the search will be sensitive to
lower/upper case of each character.
3.2
Filters
As shown in Section 1.2, the simplest way of filtering is through using the
context drop-down list within the visualization window itself when available.
It is sometimes also possible to filter the context list by selecting a context
class in the second drop-down list.
This section is about the Filters menu. Filters allow
you to quickly select multiple concepts
or relations of interest by comparing concept
and relation attributes loaded onto OVTK networks to properties you specify.
For this section and the next, we will work with a
social network rather than a biological network. We will take advantage of the
fact that Ondex is domain-independent to show some examples which are easy to
understand. Other filters and annotators will be explored in Section 5 using biological networks.
Close the Aracyc network and load a new one in: foaf.xml.gz, friend of a friend
network. The metagraph shows only one type of concept and one type of relation.
This is normal as this friend of a friend network is simply composed of
individuals who know one another.
Exercise
How many people are there
in the network?
How many relations are
there in the network?
Have a look at some of the people. Here, for example,
Marjorie McShane is observed:
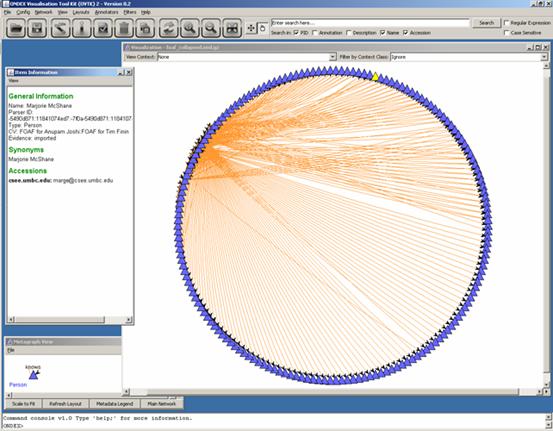
In this section, we are going to study the Shortest
Path Filter (Filters → Shortest Path). This filter is based on the Dijkstra
algorithm.
- Select one or more root concept(s).
- Select an attribute name containing
the desired relation weight, or none for a weight of 1.
- Tick directed (for a directed
shortest path) or undirected mode (leave unticked).
- Tick inverse weight if the
attribute selected is a probability rather than a weight or a distance.
The algorithm will
then compute the shortest paths from the selected concept(s) to all other
concepts. Selecting an attribute name in step 2 will result in a minimum weight
tree of the network.
We can apply the
GEM layout (Layouts → GEM Algorithm) to have a better overview of the network. In the following screenshot, we selected “Randy
Schauer” in the network.

We then applied the
shortest path filter as shown below:

The filter deleted all the
relations which were not part of a shortest path from Randy Schauer to another
person.
Note: Selecting more than
one concept will result in the network showing all the shortest paths from
these root concepts to all other concepts on the network (it will not result in
the shortest path between the selected concepts).
3.3
Annotators
In this section, we are going to study the Betweenness
Centrality Annotator in the friend of a friend network loaded in the previous
section. To do this, close the shortest path filter without saving its results
and click on Annotators → Betweenness Centrality.
You get:

By clicking on a column’s name, the table will get
sorted according to the values contained in that column. Here we can sort by
“score” which represents the betweenness centrality measure of each concept
within the network. A concept with a betweenness centrality of 1 is “central”
to the network. The table shows Tim Finin is the most popular person in this
network of friends:
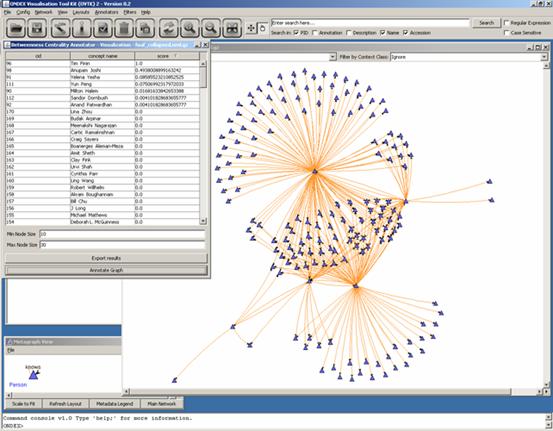
Clicking on the visualization window (after clicking
on “Annotate Graph”) will scale all concepts based on their score:

It is now easy to know what
concepts are important in the network simply by observing the size of a
concept.
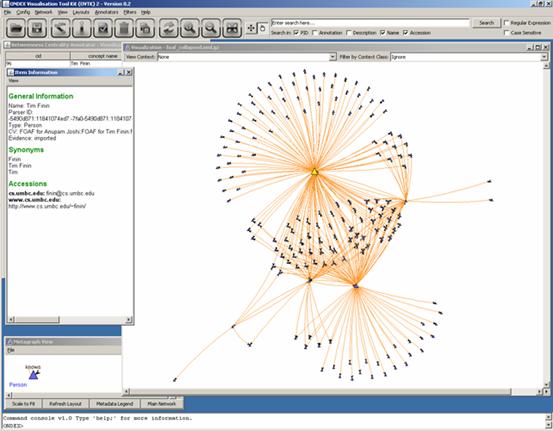
In the above screenshot,
the item information window gives details on Tim Finin. Deleting this person
would change the network the most.
4
Constructing Biological Networks in Ondex
The OVTK stands for the ONDEX
Visualization ToolKit and is used to visualize networks produced by running an
ONDEX workflow. Traditionally, the data integration is done by writing a
workflow in XML format and by running it in the back-end. We will cover this in
Section 4.2. First of all, we are going to introduce the Launcher
which allows users to prepare and run an ONDEX workflow within the OVTK. In
both sections, we are going to work with an example which will later be
analysed in Section 5.4.
For this example, techniques
of comparative genomics were combined with data integration methods to predict
pathogenicity genes in pathogenic organisms. The InParanoid algorithm[5] for the
prediction of orthologous groups of proteins was implemented as part of the
ONDEX data warehouse.
The
PHI-base database (http://www.phi-base.org/) is a reference database of virulence and
pathogenicity genes validated by gene disruption experiments. It contains
information on experimentally verified pathogenicity, virulence and effector
genes from bacterial, fungal and Oomycete pathogens identified from the
scientific literature. Data integration methods for both the contents of
PHI-base and the genome sequence of Botrytis Cinerea (http://www.broad.mit.edu/annotation/genome/botrytis_cinerea/) were developed for ONDEX.
4.1
Ondex
front-end Launcher
The Launcher is available
from File →
Launcher in the menus and looks
like this when first opened up:

In the top-left drop-down list, “All”
is selected by default which means the column below it gives all the possible
steps that can make up a workflow. If you select a type of step in the
drop-down list, the list of steps in the column below will be filtered
accordingly to only show those particular kind of steps.
All of the available steps are
documented under "Developer\Document". A simple example for a
workflow is to first select parsers (to import data) then do select mapping
methods and, finally, to select some filtering so as to improve the quality of
the graph. Any field that is not grayed out is required.
Note: Running this workflow with the whole genome sequence
from Botrytis will take about an hour. If you wish to see quick results during
this hands-on tutorial, we suggest you use a fasta file which has been prepared
for this purpose: short.fasta.
This file contains a tenth
of the entire genome and will take about 3 minutes. The results show a smaller
graph which can be analysed nevertheless. In Section 5.4, you will be able to load up the results for the
whole genome which are saved under the folder for application case 4 as
botrytis_results.xml.gz.
Note: Before running a workflow in the
launcher, make sure you save your current workflow so you can re-open it as you
might not get all the parameters right the first time around.
For this example, the steps
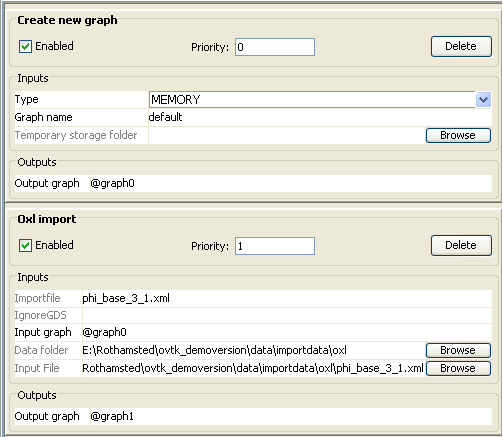
needed in the workflow are as follows:
·
Parsing the file containing the PHI-base database (Parser – Oxl import). Note: A “Create new graph” pre-step is automatically
loaded.
·
Parsing the file downloaded from the Broad institute website for the
sequence of Botrytis Cinerea (Parser –
Fasta)
Note: The taxonomy ID for Botryotinia fuckeliana was found
on http://www.ncbi.nlm.nih.gov/Taxonomy/
(40559).
CC stands for Concept Class and is therefore “Protein”.
SeqType stands for Sequence Type and is in this case “AA”, amino acid.
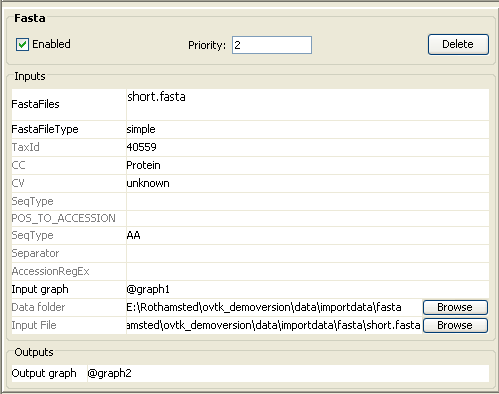
·
After parsing the two data sources, they are mapped to each other using the
“inparanoid” mapping which uses pair-wise sequence alignment results generated
by BLAST http://www.ncbi.nlm.nih.gov/blast/download.shtml
(Mapping – InParanoid).
The Evalue parameter is passed on to BLAST.
“cutoff” and “overlap” are post filter parameters which specify a bit score
cutoff and the minimum length of the match compared to the longest sequence.
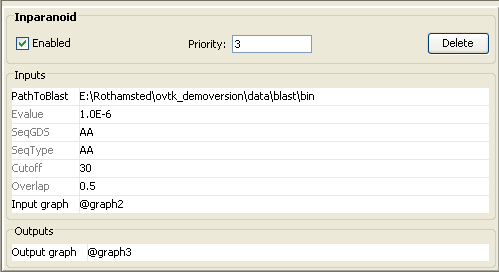
·
To reduce the complexity of the network serveral concept classes are
filtered (Filter – ConceptClass)

·
In PHI-base a protein is always linked to an “Interaction” which carries
the specific phenotype. To merge both information from protein and Interaction
the relation type set “phi1” is collapsed and the connected concepts are fused
into a single concept of concept class “Interaction:Protein”. (Transformer – Relation Collapser)
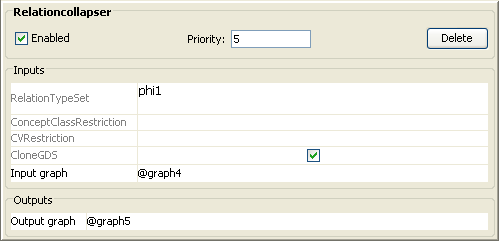
·
The unconnected filter is used to remove any isolated concepts (i.e.
without any relations to the rest of the network). (Filter – Unconnected)
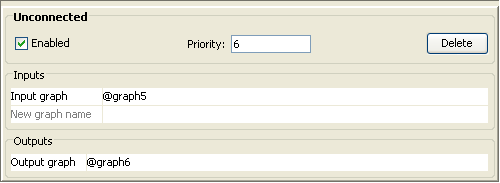
·
The next step will only keep clusters of concepts in the network which
contain at least one concept of concept class Protein from Botryotinia fuckeliana. (Filter
– Isolateclusters)
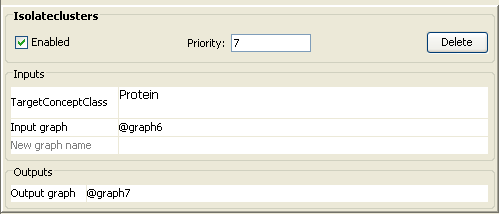
·
The resulting network is exported as OXL (Export – Oxl export) to a Gzip compressed XML file called short_botrytis_results.xml

·
Finally, a tab separated text file called short_botrytis_clusters.txt is created which lists all associated
phenotypes for the members of each cluster in the network (Export – Clusters).
Note: GDS stands for General Data Store. This is where the phenotypic
annotation is stored under the attribute name “Pheno”.

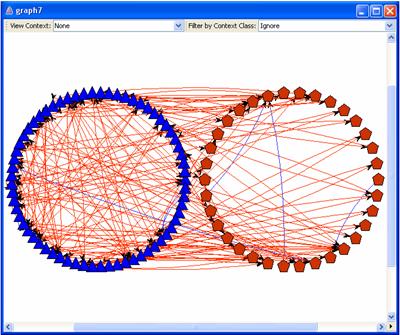
Should you wish to see the
results of your oxl network loading automatically when the workflow is finished
running, copy and paste the graph number after “Load result in OVTK when
complete” (in this case, “@graph7”). Loading short_botrytis_results.xml in OVTK will look similar to this
screenshot:
4.2
Ondex
back-end Workflows in XML
The exact same workflow could have been written in an XML file as
follows (here using the whole Botrytis genome sequence):
<?xml
version="1.0"?>
<Ondex
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"
xsi:noNamespaceSchemaLocation="ONDEXParameters.xsd">
<DefaultGraph name="phibase"
type="memory">
<Parameter
name="ReplaceExisting">true</Parameter>
</DefaultGraph>
<Parser name="oxl"
datadir="/importdata/oxl">
<Parameter
name="Importfile">phi_base_3_1.xml</Parameter>
</Parser>
<Parser name="fasta"
datadir="/importdata/fasta">
<Parameter
name="FastaFiles">botrytis_cinerea_1_proteins.fasta.gz</Parameter>
<Parameter
name="FastaFileType">simple</Parameter>
<Parameter
name="SeqType">AA</Parameter>
<Parameter
name="CC">Protein</Parameter>
<Parameter name="TaxId">40559</Parameter>
</Parser>
<Mapping name="inparanoid">
<Parameter
name="Evalue">0.000001</Parameter>
<Parameter
name="cutoff">30</Parameter>
<Parameter name="overlap">0.5</Parameter>
<Parameter
name="SeqType">AA</Parameter>
<Parameter name="SeqGDS">AA</Parameter>
<Parameter name="PathToBlast">D:/Program
Files/Eclipse/Ondex-data/blast/bin</Parameter>
</Mapping>
<Filter
name="conceptclass">
<Parameter
name="TargetConceptClass">Publication</Parameter>
<Parameter
name="TargetConceptClass">Organism</Parameter>
<Parameter
name="TargetConceptClass">Class</Parameter>
<Parameter
name="TargetConceptClass">Disease</Parameter>
</Filter>
<Transformer
name="relationcollapser">
<Parameter
name="RelationTypeSet">phi1</Parameter>
</Transformer>
<Filter
name="unconnected"/>
<Filter
name="isolateclusters">
<Parameter
name="TargetConceptClass">Protein</Parameter>
</Filter>
<Export name="oxl"
datafile="/importdata/oxl/botrytis_results.xml">
<Parameter
name="GZip">true</Parameter>
</Export>
<Export name="clusters"
datafile="/importdata/oxl/botrytis_clusters.txt">
<Parameter
name="GDS">Pheno</Parameter>
</Export>
</Ondex>
5
Customising Visualisation
of Biological Networks
Customising the way you visualise and manipulate networks is a key function of
OVTK. This is achieved through using a combination of annotators, filters,
searches and layouts.
The best way to learn about
OVTK is to try to use it on some real examples. In this tutorial we will look
at four application cases:
- Application case 1: Analysing Experimental
Data in the context of Integrated Biological Networks
- Application case 2: Functional GO
Annotation
- Application case 3: Cooking with
Protein-protein interaction networks
- Application case 4: Combining comparative
genomics with data integration for the prediction of potential
pathogenicity genes
5.1
Application
case 1: Analysing Experimental Data in the context of Integrated Biological
Networks
Author: Jan Taubert
5.1.1
Datasources
We integrated AraCyc (http://www.arabidopsis.org/biocyc/index.jsp), a database containing pathway information for the
plant Arabidopsis thaliana, with data from the former DRASTIC-INSIGHT database
for information on plant gene expression. Additionally we loaded microarray
expression data onto the concepts of the network using the General Data Store.
The expression data has been analysed for statistical significance and
normalized.
5.1.2
Objectives
We like to show, that an
integrative approach to the exploration of microarray expression data can
leverage the understanding in the context of pathways and might yield new
biological insights.
5.1.3
Steps
After loading the OXL data
file pathways.xml.gz, a meta-graph
is displayed:
11) 10) 9) 8) 7) 6) 5) 4) 3) 2) 1)

The different concept
classes are:
- Genes from the DRASTIC database, which
did not map to any AraCyc genes
- Treatments (e.g. drought,
- Merged entity consisting of Genes from
DRASTIC and Proteins from AraCyc together with microarray expression data
to achieve a more compact pathway representation
- Protein complexes described in AraCyc
- Proteins in AraCyc, for which there is no
expression data available
- Enzymes catalysing reactions from AraCyc
- Enzyme classifications for enzymes from
AraCyc
- Chemical compounds involved with
reactions and enzymes in AraCyc
- Reactions belonging to an AraCyc pathway
- AraCyc pathways which can be part of a
hierarchy
- Publications assigned to entries in
AraCyc
All eleven concept classes
can be identified in the main visualisation, which uses a circular layout. Each
circle corresponds to one concept class, e.g. the circle with green round
concepts on the left corresponds to Treatments from DRASTIC, whereas the circle
with orange star shaped concepts corresponds to Pathways from AraCyc.
The whole network contains
24541 concepts and 45153 relations. Using context information we can display
only the relevant sub network instead of having to work with the whole network.
From the Context drop-down
list please select gamma-glutamyl-cycle.
The resulting network has still the circular layout from the whole network.
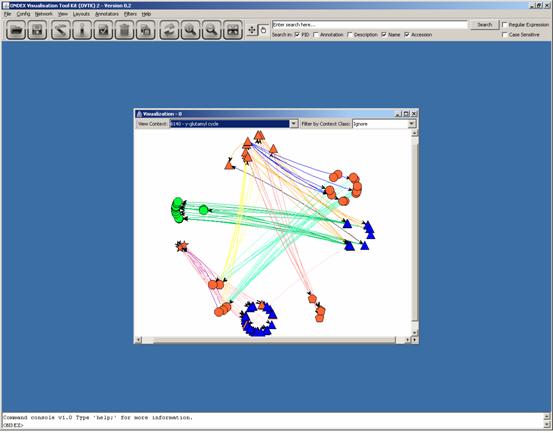
The GEM layout (Layouts -> GEM Algorithm) can easily be applied to this smaller network, which
produces a more pleasant representation of the network.
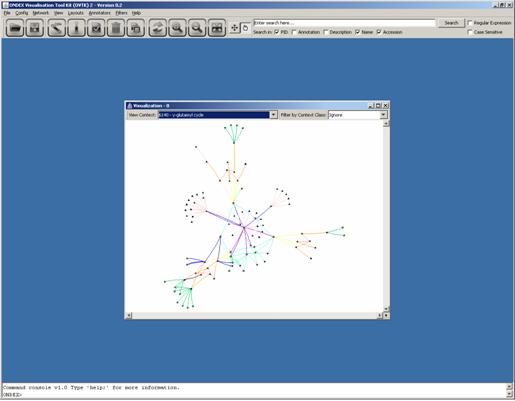
Now additional features
like displaying concept and relation labels (View -> Show Concept Labels, View -> Show Relation
Labels) and anti-aliased painting (View
-> Anti-aliased) can be turned on
the enrich the current visualisation. Using the Mouse wheel you can zoom into
the network view. Try to locate a group of membrane
alanyl aminopeptidases.
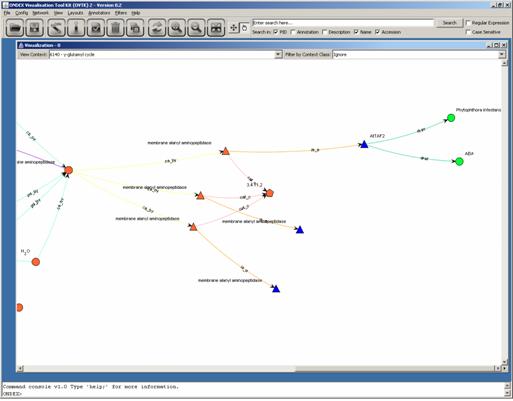
Here only one Gene (blue
triangle) has information associated from the DRASTIC databases (green circle).
On all of these three genes additional information can be displayed by right mouse click on the concept.

By clicking on Edit Concept Properties you are able to
inspect all the properties assigned to this concept. Selecting View/Edit Concept General Data Store
displays a tab based representation of all values in the General Data Store.
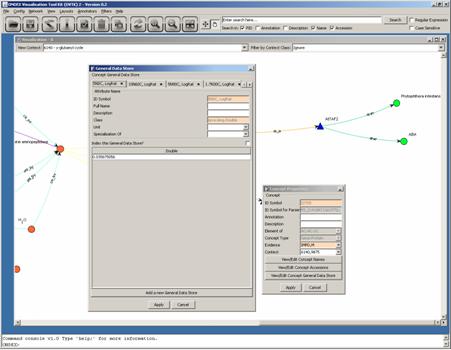
In this case the values in
the General Data Store are the microarray expression data listed according to
the treatment. Now we can use the Scale by Value annotator (Annotators -> Scale by Value) to actually map the values of the General Data
Store to the visualisation of the corresponding concepts.

The annotator supports
multiple value selection. Here all treatments of the form xN60C are selected. Annotate
Graph will perform the changes to the visualisation.
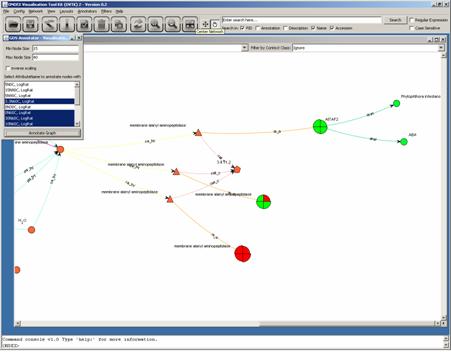
The visualisation for the
concepts now changed to pie charts. The pie chart is divided into the number of
treatments that were selected in the annotator. The order of the pies is in
mathematical positive direction (anti-clockwise), e.g. the 3.3N60C treatment
expression value is the upper right pie. Visual inspection of this network now
reveals, that Gene in the middle shows an irregular expression pattern, whereas
the other two Genes are oppositely regulated (red for up-regulation and green
for down-regulation).
Further inspection of the
value in the General Data Store supports the hypothesis that this measurement
might be wrong and the Gene is behaving in the same way as the other
down-regulated Gene.
In addition we can annotate
one of these three Genes with information from the DRASTIC database showing
that this particular Gene has a significant change in expression under the
associated conditions.
5.1.4
Summary
In this example we showed
how visualisation of expression patterns can leverage the understanding in
terms of comparing conditions and influenced pathways. We identified one
particular Gene with an irregular expression pattern. Furthermore data
integration helped to enrich pathways from AraCyc with information about
influential treatments from DRASTIC.
5.2
Application
case 2: Functional GO Annotation
Author: Keywan
Hassani-Pak
5.2.1
Data
sources
In this application case we are going to show the
integration of major public databases to provide a comprehensive gene
annotation network. We have parsed and integrated GOA EBI[6],
UNIPROT[7],
MEDLINE[8]
and Gene Ontology[9]
step by step in the ONDEX backend to build a core database. In this example we use
the GO annotations of Arabidopsis
thaliana, however this core database can be created for any species. From
the
5.2.2
Objectives
The functional annotation of
genes is still a major challenge in the post genomic era. Traditional manual
annotations by literature curation are reliable and of high quality. However,
as both the volume of literature and of genes requiring characterization
increases, the manual processing capabilities are becoming overloaded. To
efficiently annotate genes with controlled vocabularies such as Gene Ontology
(GO), computational methods to automate the process of functional annotation
are required. We are working towards a novel annotation system for ONDEX that
includes data integration and literature analysis methods to predict the
function of previously unannotated genes.
Our first aim is to create a
comprehensive integrated network in which genes are enriched with manual (if
available) and automatic annotations such as text mining based annotations. The
second aim is to show the information content of the core database and how to
navigate through the network gene by gene.
5.2.3
Steps
The data integration was done in the ONDEX backend.
Some intermediate steps were exported into the OXL format. Here we are going to
load these steps and show some examples. As the first step we load the GOAEBI_MANUAL.xml.gz file into OVTK.
The Metagraph View is displayed. We
can see that Proteins are connected to the three GO classes (Molecular
Function, Biological Process and Cellular Component). In this case every
relation has a publication assigned to it (ternary relation). The publication
contains the evidence for the relation between Protein and GO. Clicking on the
Metadata Legend button shows some information about the number of the concepts
in this first graph. For example 1157 unique publications are referenced in the
GOA Arabidopsis file.

Let’s search for “at2g” to get a list of genes located
on the second chromosome of Arabidopsis. Select several of the results and
apply the Neighbourhood Filter with a
depth of 1 or 2. Now we open the main graph and apply the GEM Algorithm Layout. We can see a graph with several clusters,
which shows GO concepts surrounded by associated proteins.
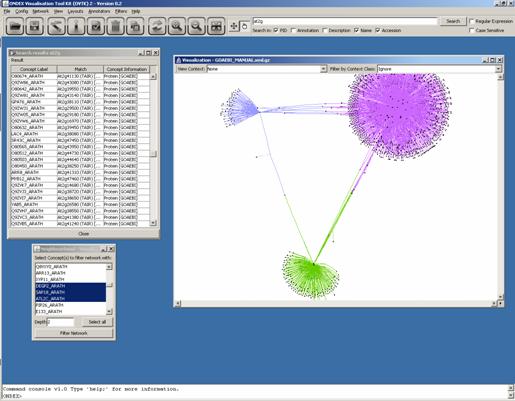
The proteins have no sequence or further details right
now. However they contain UNIPROT accessions, which we will use for the next
integration step. Now let us load GOAEBI_MANUAL_UNIPROT.xml.gz
and have a look at the Metadata Legend.
We see that the number of Publications has increased to 2446 and 199 new EC
concepts are in the graph now. This is because UNIPROT proteins have their own
publications and ECs.

We perform the same accession based integration
approach with MEDLINE and GO accessions. The results can be loaded from GOAEBI_MANUAL_UNIPROT_MEDLINE_GO.xml.gz.
This file is an integrated core database which contains sequences, abstracts
and the GO hierarchy. It can be used for many automatic GO annotation
approaches as a manual reference set.
Now we are going to show on some example proteins the
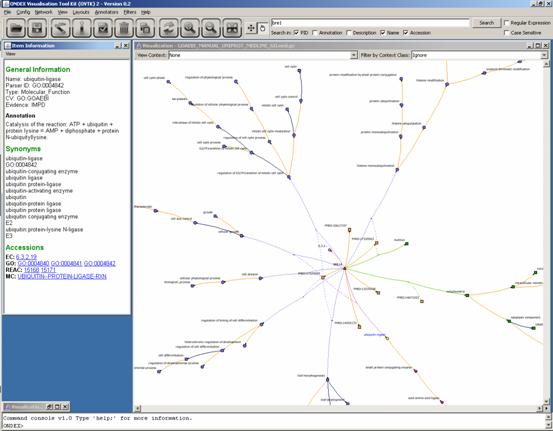
information content of the integrated gene annotation network. First we search
for “bre1” and apply the Shortest Path Filter. This shows the
protein in the centre with its publications and GO annotations. Additionally
the GO hierarchy is visible.
Next we search for “bik1” and apply the Neighbourhood
Filter with depth 1. Then we open the Item
Information window and click on one of the publications. The title,
abstract, year and journal etc of the publication are shown in this window.
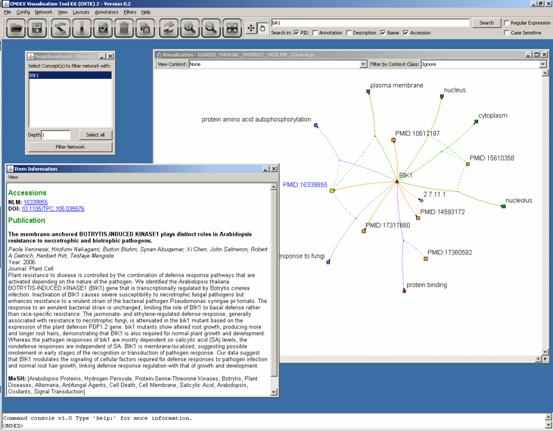
As we can see, not every publication was used as a
source for functional GO annotation. We are now interested to extract automatically
from these publications some GO terms. This text mining based mapping between
publications and GO concepts was carried out in the ONDEX backend. We load the
results into OVTK:
GOAEBI_MANUAL_UNIPROT_MEDLINE_GO_TM_noHiearchy.xml.gz
Search for “mgp”
gene and apply the Shortest Path Filter.
The blue relations (is_r) are created automatically by the text mining mapping
method. A score and evidence sentences are assigned to each of those relations
(can be seen in the Info Window).
Applying the “Scale relation width by
value” annotator will highlight strong relationships.
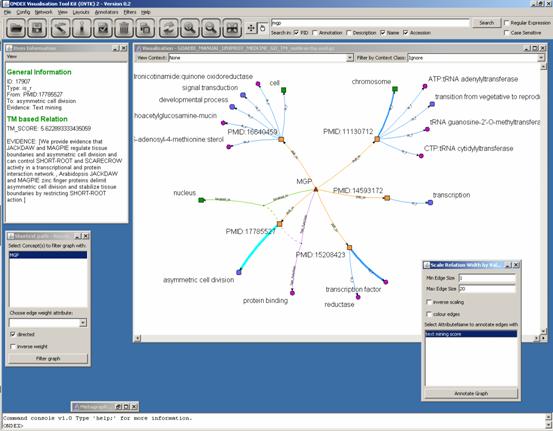
We perform the same search with “FH8” and apply the Neighbourhood
Filter with depth 2. One can see on the right side that one publication (PMID:11130712)
has lots of links to proteins, it seems to be a large scale genomic DNA paper.
On the left side manual and automatically extracted GO annotations can be
compared.

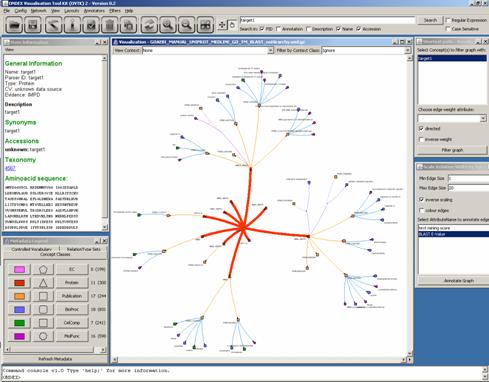
As a last step of the analysis we have blasted two
target sequences against our integrated database. So let us load GOAEBI_MANUAL_UNIPROT_MEDLINE_GO_TM_BLAST_noHirarchy.xml.gz
into OVTK. We search for “target1”
and apply the Shortest Path filter again. Then we annotate the graph relations
by E-Value in Scale Relation Width by
Value. We can see our target protein in the centre connected to several ARR
proteins based on sequence similarity.
5.2.4
Summary
We have integrated major
public databases into one core database. OVTK enables a sufficient way for the
graph based navigation of these gene annotations networks. This application
case showed that integrated gene-annotation networks can provide substantial
support for semi-automated genome annotation projects. Using text mining as an
automatic GO annotation method opens up the wealth of indispensable knowledge
in the scientific literature.
5.3 Application case 3: Cooking with Protein-protein interaction networks
Author: Jochen Weile
5.3.1 Context
This application case uses data
from an ongoing project that examines topological comparison on protein-protein
interaction networks (PPIs) with the help of graph hierarchies that serve as
scaffolds for the comparison.
5.3.2 Objectives
·
We will have a look at the single interim steps of
the workflow to understand the whole process as well as the structure of the
graph.
·
We will learn about the OVTK’s abilities to serve
as a tool for quality assessment and data exploration.
5.3.3 Data sources and processing workflow
Integrated databases: We
integrate several databases during the time of the workflow: PPI data from a
high-throughput YTH-array experiment on Campylobacter
jejuni[10],
the respective PPI datasets for E. coli
and Helicobacter pylori from the
IntAct database[11],
the Pfam-A HMM library[12]
and the Gene Ontology[13].
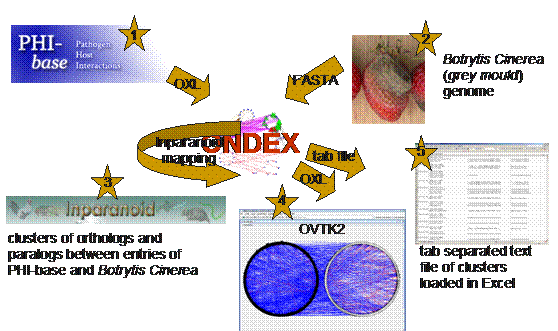
See fig. 5.3.1.

Figure 5.3.1
General Overview of the data integration and processing workflow in this
tutorial.
5.3.4 Steps
Step 1 (Integration of PPIs): Let us have a look at the
workflow that was used to create the data for this tutorial. Open the workflow
launcher (File→Launcher) and load the file DemoWorkflow.xml
(File→Open). Do not execute it, though. It
requires special hardware and takes several hours to run. Anyway, we can now
have a look at the single integration and processing steps in it.
·
PSI-MI parser: The PSI-MI parser imports the PPI
data from IntAct and from the C. jejuni
experiment flatfiles. It creates concepts for each participant of an
interaction and links them with relations. The relation types match the
description of the interaction. It may be a general “interacts with”, or more
specifically “physical interaction”, “colocalization” or “is part of”.
·
Uniprot2GOA transformer: This transformer uses the
Uniprot IDs found in concepts with a given Taxonomy ID (in this case 83333,
that is E.coli) to find the
corresponding GO annotations in the GOA database. It then attaches these
annotations to the concepts.
So we now have our interaction networks
with annotated E.coli entries. Let’s have a look at it! It has been exported to
Step1.xml.gz. Switch back to main OVTK window and open the file
(File→Open).
After a little wait (~60,000
relations are being loaded) the metagraph window will appear. The metagraph
gives an overview of the main graph representing every concept class as a node
and every relation type that connects part of the concepts in two concept
classes as an edge. The real graph visualization is minimized at the lower left
corner of the main window. I would not recommend opening it though, since it
would take some time to render all those 60,000 relations. For starters it will
be enough to examine the metagraph.

Figure 5.3.2: Metagraph at step 1 of the
workflow.
If you right-click on nodes or
edges you will see a context menu popping up. It explains the represented type
and the number of associated elements in the main graph. In the example in
fig.5.3.2. this would be the type RNA that represents 2 concepts. The menu also
offers to change the visibility of these elements in the main graph.
You will probably be wondering
now, why there are other concept classes than “Protein” in the graph. That is a
legitimate question. There is a quite simple answer: The E.coli dataset from IntAct did not only contain entries about
protein-protein interaction, but also about other kinds of molecular
interactions. Since we do not need them here, however, we might just as well
get rid of those entries.
Step 2 (Filtering the data): To do so
in the OVTK we would make those data points first invisible and then remove all
invisible elements from the graph. Right-click on all the metaconcepts (nodes
in the metagraph) except “Protein” and then click their visibility checkboxes
in the appearing context menus. You will see that this does not only affect the
metaconcepts themselves, but also their connected metarelations. This is
because relations cannot exist without a source or target concept.
Now all unnecessary elements are
invisible we can remove them from the graph. This can be done by the graph
synchronization function (Network→Synchronize Network).
However, since it would take quite a long time on such a big graph (~30 min.),
because the consistency with the visualization would have to be maintained all
the time. Luckily there is a quicker way: The ONDEX workflow.
Bring the Launcher window to the
front again and have a look at the next elements in the workflow:
- Concept class filter: This filter removes all
concepts from the graph that are of one of types given in the list. Here:
“Thing, Reaction, DNA, sDNA, RNA, SMOL”.
- Relation type set filter: This filter removes
all relations form the graph that are of one of the types given in the
list. Here: “it_wi, is_p”.
- GDS value filter: This filter preserves only
those concepts that feature one of the given values for the selected
attribute. Here: “197 (C.jejuni), 83333 (E.coli), 210 (H.pylori)”.
- Unconnected filter: This filter removes all
concepts that do not participate in relations any more.
So all we need to do to see the
results of this cleanup is to switch back to OVTK and load the file Step2.xml. We
want it to look nice when we open it later, so we apply a layout to it: The GEM
layout (Layouts→GEM Algorithm). This will take a while
so let’s move the progress monitor some place where it doesn’t obstruct us and
do something else in the meantime.
Start the statistics module (Network→Statistics). You see a window that is
divided into three main areas. Top left is the listing panel. It contains three
lists with different graph elements that can be used as variables or filters
for the module. Top right is the selection panel. It contains the variable
field with two buttons that load it with the currently selected element or
unload it, respectively. Below the variable field you can see the filter list
with the same kind of load/unload buttons beside it. The filter field can be
loaded with an arbitrary number if elements, that will be handled as conditions
linked with an “or” operation. If attribute names are loaded into the filter
list they may be assigned a value.
Finally, the panel on the bottom
is the display panel. It shows the number of concepts that were found to meet
the selected conditions, the respective number of relations. In case the
variable can contain numbers, it also features the mean and the standard
deviation values, and a histogram.

Figure 5.3.3: The
statistics module.
This probably all sounds quite complicated so let’s just
try it out and see an example. Select the attribute “CONF” from the listing
panel and load it into the variable field by clicking the respective button.
You should now see a histogram of the confidence values that occur in the
graph. Most of the interactions seem quite unlikely, with the highest peak at
0.1 and the mean value at 0.3354. This seems to confirm that YTH is a very
hypersensitive methodology.
You can also see the number of relations that hold
confidence values: 11,900. This is a surprisingly small number, considering
there are about 60,000 relations in the whole graph.
So let’s find out where they come from. Unload the
variable again and select the “TAXID” attribute. Load it into the filter field
and assign the value “197” (i.e. C.jejuni).
Now load the confidence attribute into the variable field again.
The histogram seems unchanged but we see that the number
of relations is a bit lower: 11,885 Does that mean there are only 11,900 –
11,885 = 15 relations with confidence values in the whole rest of the dataset?
That would explain why the histogram looks almost the same as before. Let’s
find out.
Unload the variable, change the TAXID value to 83333 (i.e.
E.coli) and reload the confidence
attribute into the variable field.
Indeed, there are only 15 relations: Six with value 0.4,
five with value 0.6 and four with value 0.8.
That would mean that the H.pylori data does not contain any confidence values. Find out; you
know what to do (H.pylori-TAXID=210).
The commonality between the E.coli and the H.pylori
dataset is that they both originate from the IntAct database while the C.jejuni data comes from the
experimental flatfiles. It would seem that the IntAct dataset is of quite poor
quality in these cases.
Anyway, the layout process should be finished by now. If
it is, we can have a look at the graph now. However, in order to not wait for
too much rendering we should hide the relations first. Open the metagraph
window again and right-click on the “physical interaction” metarelation.
Unselect the visibility checkbox in the context menu. If you think your machine
is fast enough you can also omit this step, but be prepared for some 30 seconds
of GUI freezing. Now de-iconify the main visualization window (lower left
corner) and click the “center network” button in the toolbar. There they are:
two black spots. These are the two main components of the C.jejuni and the E.coli
PPIs. Scattered around them you can see some fragments, among them the small
set of H.pylori data.
To see the differences between the species we could apply
different colors to them. Open the Color Annotator (Annotators→Color by Value). Select the “TAXID” attribute from the list and click on “Annotate
graph”. A legend will appear, explaining the color code. (You will probably not
see any change from the current zoom level unless you have activated
anti-aliasing, which I would not recommend at the moment).
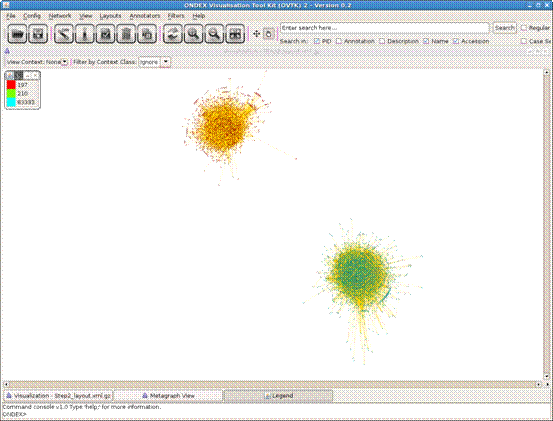
Figure 5.3.4: Two
interaction networks in GEM layout colored by Taxonomy ID.
Step 2.1 (Examining
a subgraph): Zoom
in on the C.jejuni main component
(the less dense one): Hold down the shift-key and draw a rectangle over the
component to select it. Then click the “Zoom in” button on the toolbar.
Alternatively you can use the mouse wheel.
We can see the effects of the graphs scale-free property:
Many nodes with low degree attached to few nodes with high degree. But to see
this a bit clearer we could have a look at a smaller subset of it. To this end
we pick a node that gives a nice example: The “CRISPR-associated protein”. The
easiest way to do so is to use the search function. Enter “crispr” in the
search field (top right) press enter (or click search). A list appears that
shows the result. Select it and you will see the node being highlighted in the
graph.
Now open the neighborhood filter (Filters→Neighborhood),
specify search depth as ‘3’ and click “Filter Network”. You can now apply the
GEM layout again; it will be much quicker on a smaller graph like this. You may
also active anti-aliasing now if you wish.
In order to examine this subgraph a bit more we would have
to synchronize it first. But to spare you the waiting time I prepared the
result for you. You can close all internal windows and open Step2_mini1.xml.gz.
Let’s find out more about the internal structure of this subgraph.
Choose the ‘Betweenness centrality’ annotator from the
menu (Annotators→Betweenness Centrality). Wait a
few seconds for it to compile the required data; then a window will open that
shows a list of the nodes in the graphs with their respective BC scores. Click
on the score category to sort them and then click on the entry with the highest
value. It will then be highlighted in the graph. Click on “Annotate Graph” and
minimize the window.
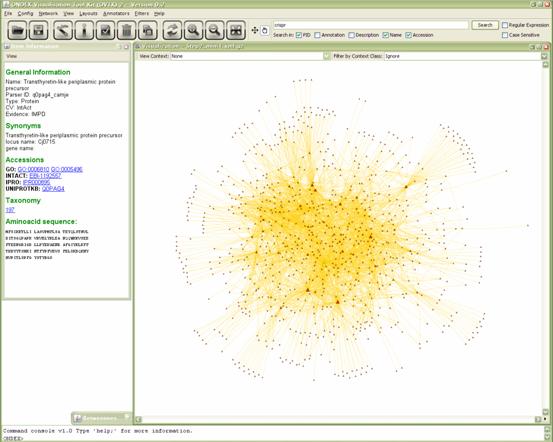
Figure 5.3.5:
Betweenness centrality filter applied to a smaller subset of the graph.
You will see that a few nodes are scaled larger then most
of the others. Those are the ones that feature the highest BC score. To learn
more about them click on the info button in the tool bar and select one of
them.
If you like, you can also save the results of the analysis
in an Excel spreadsheet. De-iconify the annotator window and click on “Export
results”.
Step 2.2 (Examining
a small subgraph): By
applying the Neighborhood filter again with a lower depth (2) we can get an
even smaller subset. This time we can call the synchronization for real. It
will only take about a minute. Afterwards we can apply the GEM layout anew. Now
we can see clearly the scale-free property of the graph. Many low degree nodes
associated to very few high-degree nodes.
Our graph is now small enough to run a more complex
analysis: The “Virtual Knock-out” annotator (Annotators→Virtual Knock-Out). After a little wait ( O(n3)
) another internal window will appear showing a table with the results for
each node. It contains the following columns:
- Node
ID
- Node
name
- Extension:
The number of paths that were extended if a node was added.
- Deletion:
The number of paths that were deleted if a node was deleted.
- No
change: The number of paths that would not change if this node was
deleted.
- The
score computed for this node.
- Diameter:
The length of the longest efficient path in the subgraph that
contains the node.
- Components: The connected components that
the graph falls apart to if the node would be removed.
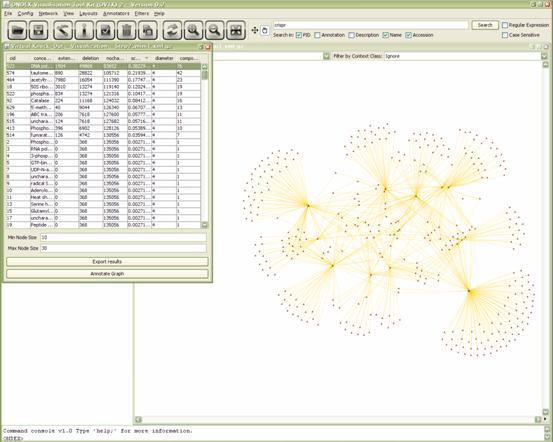
Figure 5.3.6:
Knockout filter being applied to a small subset of the PPI.
Just like the Betweenness-centrality annotator you can
sort the entries by their score, select the highest entry and click on
“Annotate Graph”. Then minimize the window to see the effect on the graph; or
you might export the results to an Excel spreadsheet again.
You can use the item information window again to explore
the graph a bit.
To find out more about the significance, we can apply some
more filters. Let’s try the shortest path filter, select our central CRISPR
protein again and open the shortest path filter (Filters→Shortest Path) choose the confidence attribute as edge weight. Since the confidence
is a probability value we have to inverse its value (so lower probabilities
result in a greater distance value). Now click “Filter graph”.
We can see that a lot of paths that seemed visually longer
were actually considered improbable.
There is a nice layout trick that we could apply now:
click “Re-layout” on the toolbar, close the shortest path filter. You will be
asked if you want to keep the changes of the filter; click “No”. You will no
see the complete subgraph laid out according to the shortest path tree
structure. (See fig. 5.3.7)
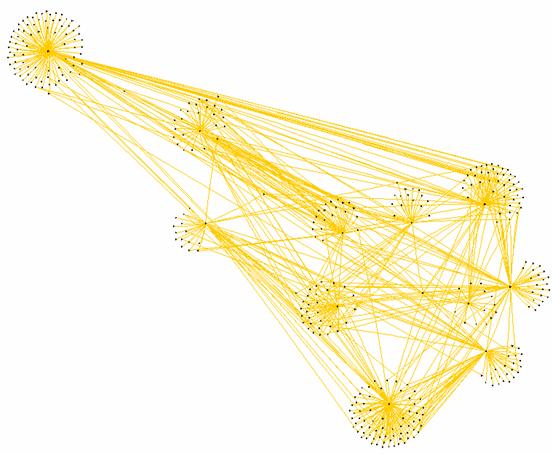
Figure 5.3.7:
Subgraph around the CRISPR protein laid out according to its shortest path
structure.
If you want to you can try the same with the all-pairs shortest
path filter. (Filters→All pairs shortest path) But it
might take some time, since it is another O(n3)
algorithm. It does the same as the shortest path filter, except it does not
only consider one source node but computes all shortest path between all
possible pairs of nodes.
Let’s try some more effective filter for significance
analysis. The significance filter (Filters→Significance)
Select the confidence attribute and choose a threshold
value in the chart. Then click Filter graph.
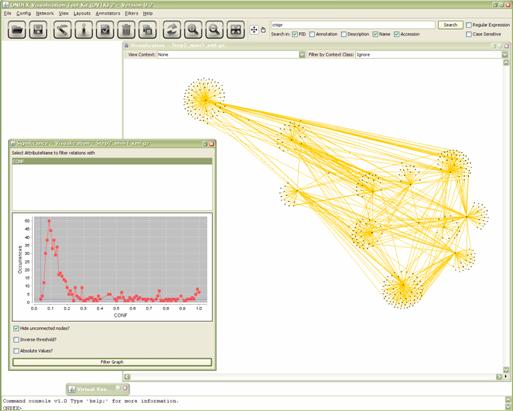
Figure 5.3.8:
Subgraph before application of the significance filter.
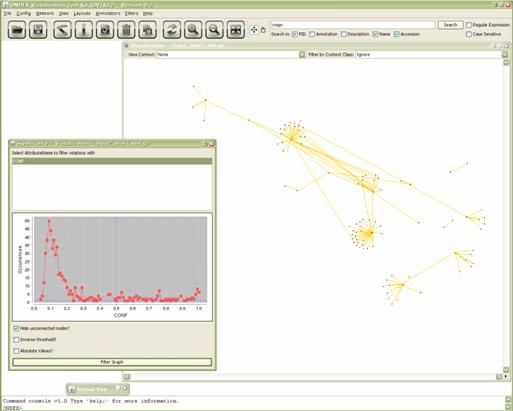
Figure 5.3.9:
Subgraph after application of the significance filter
We can see that a whole lot of interaction relations being
removed. This is actually not surprising because the histogram already suggests
that most of the interactions have a confidence value below 0.3. To see how
this affects the complete graph. You can load Step2.xml.gz again and apply the significance filter to it.

Figure 5.3.10: Complete PPI of C.jejuni
after application of the significance filter.
Since we found out earlier that neither the E.coli nor the H.pylori data features confidence values we already knew that this
filter would not affect those two datasets. However it might still be a good
idea to apply it in the workflow as well. Switch back to the launcher and you
will see that the next step is indeed:
- Significance
filter: removes all elements from the graph that feature a value lower
than the given value (here: 0.5) in the given attribute (here:
confidence).
Step 3 (Seed
mapping): If we
look further on in the workflow we see the next steps that are part of the
Pfam-based seed mapping, which works as follows:
- Inparanoid[14]
mapping: This algorithm uses BLAST to compute possible paralogs and
orthologs between the three proteomes in our graph and represents them as
relations of the respective type. We low e-value and score cutoffs (10-2,
50) resulting in a very sensitive scan, but also in a great number of
false positives.
- Relation
type filter: This filter removes all relations of the selected type (here:
paralog). We are only interested in the orthologs, so we can just as well
get rid of the paralogs.
In the next step we try to get rid of those false
positives by trying to confirm each orthology relation with common protein
family hits. All orthologies that fail to present at least one shared Pfam
module are removed. Also, to proceed as conservatively as possible, we have a
look at the protein’s GO annotations (where existent). If they differ too much
between the two ends of an orthology it is deleted as well. To this end we need
a graph configuration as it can be seen in fig. 5.3.11.

Figure 5.3.11: Metagraph of the configuration used for the Pfam-based
seed mapping.
Hence the next steps in the workflow are:
- Pfam
parser: reads the Pfam database and creates a concept for each domain and
family.
- Sequence
to Pfam mapping: Uses HMMer to find the protein families and domains for
all proteins. It then creates relations of the respective type.
- Pfam-based
ortholog filter: This filter is the actual core of the method. It performs
the checks explained above to each orthology relation.
- Concept
class filter: After the seed mapping is done we do not need the Pfam data
anymore.
Now, let’s have a look at the graph after the application
of the seed mapping. Bring the OVTK to front again, close all internal windows
and open Step4_layout.xml.gz. In the metagraph you will see
that there is a new relation present in the graph:
To spare you waiting for the layout process, I already
saved one in the general data stores of the concepts for you. Just choose Layouts→Static to load it. Then hide the
“physical interaction” relation using the metagraph context menu. You may then
de-iconify the main graph window.

Figure 5.3.12: The Two PPIs of E.coli and C.jejuni with orthology
mapping.
On the first glance it looks quite good. Let’s have a look
at the statistics. Open the Statistic module once more. The scores for the
mappings are also stored in an attribute called confidence. Hence we have to
make sure, that we only include orthologies in our statistic. Load the relation
type “ortho” into the filter panel. Then load the “CONF” attribute into the
variable field. You will see something like the contents of fig. 5.3.13.
There are no orthologies with a score lower than 50. This
is, because we chose 50 as our cutoff for the Inparanoid algorithm. The
histogram rises between scores 50 and 100. This seems reasonable, since we can
expect that the probability to share a Pfam hit rises with increasing sequence
similarity. From score 100 on, we see the number of occurrences falling again;
obeying the typical blast hit score distribution.
In total we see a number of 948 orthology relations. We
must not forget that this is based on bi-directional BLAST hits, so we have two
relations for each orthology. So we have 948/2 = 474 actual mappings. How much
is that compared to the size of our C.jejuni
dataset? To find out, empty the fields again and load the “TAXID” into the
filter field. Assign value 197 for C.jejuni.
Then load the concept class “Protein” into the variable field. And, the answer
is: 1321. So we have a share of 474/1321 = 0.359 = 35.9% of our C.jejuni proteins mapped to E.coli and H.pylori proteins.
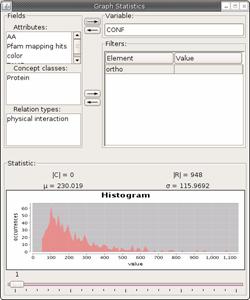
Figure 5.3.13: Statistics module showing a histogram of the BLAST scores
in the orthologies.
Step 4 (Hierarchy
construction): In
the next step we build up a clustering hierarchy on our PPIs. This is supposed
to work as a scaffold for the comparison that is to be made between the PPIs.
Have a look at the workflow again.
- Hierarchy
building transformer: This transformer uses the methods invented by
Clauset et al.[15]
to create a clustering hierarchy each single PPI in our graph. This is
based on the idea that a network can always be seen as a cluster of
sub-clusters of sub-sub-clusters... and so on.
Let us have a look at the result. Load the file Step5.xml.gz. We see that the metagraph has
changed again. Now we have hierarchy nodes that are connected to themselves and
to the proteins with “is part of” relations. The Hierarchy is basically a tree,
so we could use a tree layout to display that graph. But first we have to make
sure that the interaction and mapping relations don’t interfere with that tree
structure. So we just hide them with the metagraph.
Now we can apply our tree layout (Layouts→Tree).
However our tree structure is based on “is part of” relations instead of
“includes” relations. So we have to reverse the order. Click Layouts→Layout Options and check the box for “reversed
edge direction” in the appearing window, then click “Refresh Layout”. Now you
may de-iconify the visualization window.
What may appear like an obscure line are the two tree
structures. You may see if you zoom in on it.
Now we will try to visualize the mapping between the two
proteomes again. Zoom in on the gap between the two trees and start marking the
smaller one from there. The “Color by value” annotator might help you find it.
When you have marked the tree use the flip layout (Layouts→Flip) to reverse it and then drag it
below the other tree. No we can set the orthology relations to visible. Use the
metagraph to do so.
You may explore the graph a bit using the item information
panel.

Figure 5.3.14: The two PPIs with their hierarchies and their seed
mappings.
You might also have a look at a smaller example. Close all
internal frames and load the file Step5_mini.xml.gz. Again I have prepared a layout for you. You can load it
again using the static layout.
Figure 5.3.15: Small subgraphs of two PPIs with their hierarchies and
seed mappings.
You may explore the graph a bit using the item information
panel or any annotators.
The last step in this procession workflow would of course
be the actual topological comparison. It is, however, still under development.
Anyway I hope you have gained some feeling for the idea behind this project
during this tutorial.
5.3.5 Summary
In this tutorial we have learned
about different visualization and data exploration possibilities in the OVTK.
We have also begun to understand the basics of the graph data structure of
ONDEX and the way the workflow engine can extend and alter it.
Finally, we have understood the different steps that are
made in ONDEX’s PPI comparison project.
5.4
Application
case 4: Combining comparative genomics with data integration for the prediction
of potential pathogenicity genes
5.4.1
Datasources
We integrated PHI-base (http://www.phi-base.org/), a database
containing expertly curated molecular and biological information
on genes proven to affect the outcome of pathogen-host interactions.
Additionally we loaded the genome sequence of Botrytis Cinerea (http://www.broad.mit.edu/annotation/genome/botrytis_cinerea/).
5.4.2
Objectives
Running our implementation
of the InParanoid algorithm based on BLAST mappings of the genomic data gives
us new biological insights. Indeed PHI-base contains a lot of annotation which
can be displayed on the network using the “Annotators” menu. This type of
visualization in OVTK allows us to gain new hypotheses on the Botrytis genome.
5.4.3
Steps
After loading the OXL data
file botrytis_results.xml.gz, a
metagraph is displayed. The metagraph shows red circles are genes from Botrytis
and blue triangles are genes from PHI-base (see explanation for
“Interaction:Protein” Section 4.1).
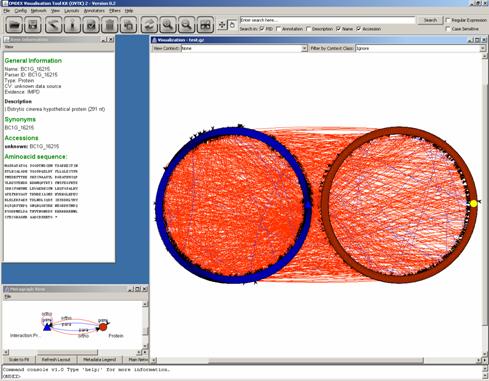
They are linked by ortholog
and paralog relations indicating whether genes were separated by the event of
speciation (ortholog) or genetic duplication (paralog). Orthologs retain the
same function in the course of evolution, whereas paralogs evolve new
functions, even if these are related to the original one.
Concepts imported from
PHI-base contain a lot of annotation. In order to make this annotation visible,
we are going to use the Annotators
menu. First of all, let us use the Colour
by Value annotator.
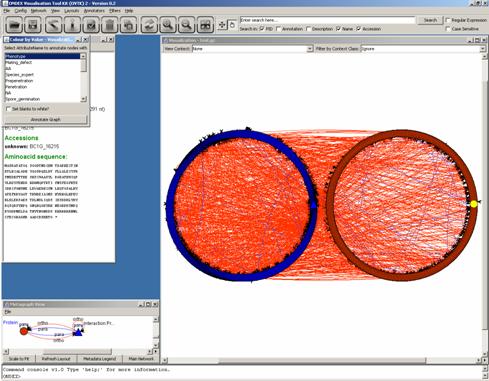
As a lot of phenotypic
information is encoded in PHI-base, we select the first attribute: “Phenotype”.
Then click on “Annotate Graph”. We get a colour legend and colour annotation on
the triangles (PHI-base concepts) in the graph:
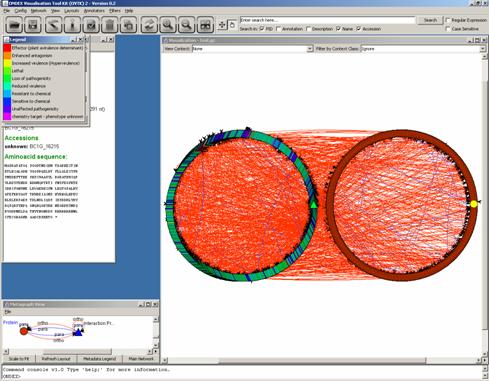
We use Layouts
→ GEM
Algorithm in order to visualize
clusters:
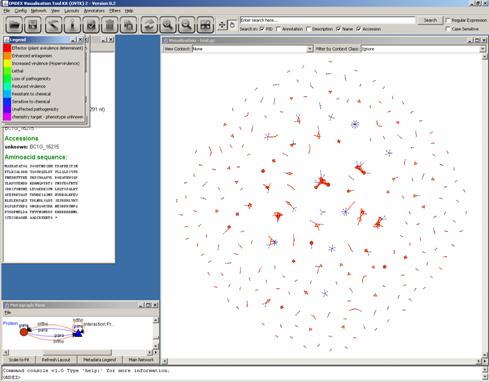
We may now zoom in on a
particular cluster:
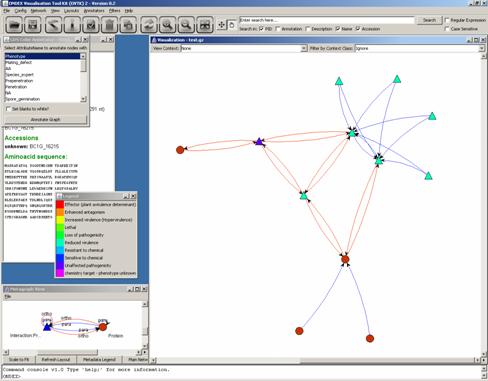
Another Annotator we may use is the Shape by Value Annotator as PHI-base
also contains information on the pathogen species. We select the attribute
“TAXID” (Taxonomy Identifier) in the list:
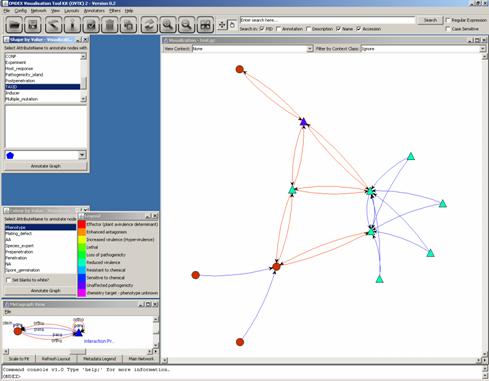
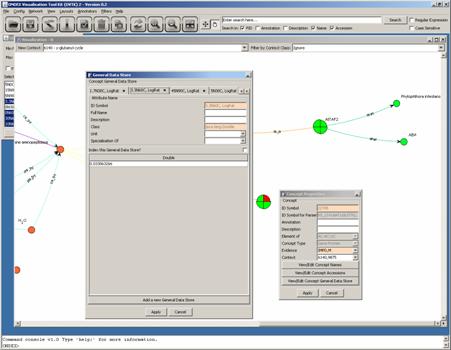
We can look up the
concepts’ TAXID by editing their concept properties:
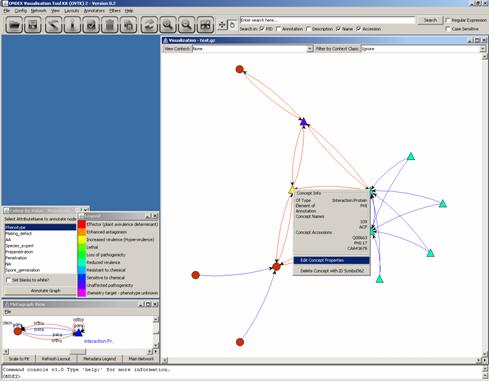
By clicking on “View/Edit
Concept General Data Store”, we get:
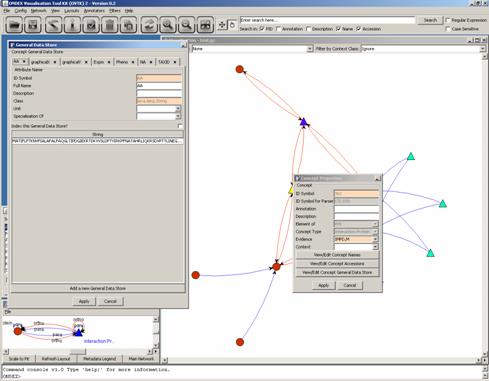
The taxonomy ID is under
the TAXID tab. When there are a lot of tabs, you may have to click on a right
hand side arrow to get to it.
Once you know what TAXID a
concept holds, you may enter it in the box below and choose a shape you would
like to associate to it:

The results are displayed
once you click on the graph:
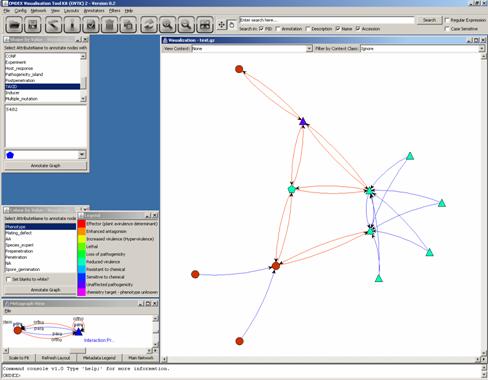
Let us change the shapes of
all the PHI-base concepts in this cluster by using the same annotator several
times. (Note: Entering more than one TAXID in the blank box would make the same
shape be associated to all of those TAXIDs.)


5.4.1
Summary
In this example we have
shown how data integration combined with visualization of annotation from a
manually curated database can yield new hypotheses for a genome.
6
Technicalities
6.1
Availability
OVTK is open source and the
latest nightly build can be obtained from: http://www4.rothamsted.bbsrc.ac.uk/ondex/latest_snapshots/ovtk2_packaged-distro.jar
6.2
Runtime
environment
OVTK requires a JAVA 6
runtime environment (http://java.sun.com/). It can be used under both Windows and Linux. You can
execute the downloaded file using the following command:
java –Xmx512M –jar
ovtk2_packaged-distro.jar
The parameter –Xmx512M is used to specify the maximum amount of memory JAVA
can use. The minimum amount for OVTK is 256 MByte. The more memory is
available, the more OVTK can cope with bigger networks.
During the first run
several resources are created. These include two directories, “data” and “net”.
The “data” directory contains examples, help files, configuration files and
images used by OVTK. The “net” directory is part of the scripting engine of
OVTK, which also created the corresponding help file “Scripting_ref.htm”. Last
but not least a log file called “ondex.log” is created.